

Las enfermedades congénitas del aparato respiratorio comprenden un extenso número de patologías del desarrollo embrionario que pueden comprometer el desarrollo de la laringe, la tráquea, los bronquios, el parénquima pulmonar, los vasos, el diafragma o la pared torácica. La ecografía prenatal ha hecho posible que un gran número de malformaciones pulmonares sean ya conocidas antes del nacimiento. Las técnicas de imagen (radiografía, ecografía, tomografía computarizada) son esenciales para el diagnóstico. El tratamiento es quirúrgico en la mayoría de los casos. Estudiaremos la embriología, las alteraciones del desarrollo pulmonar, la malformación adenoide quística, el secuestro, el quiste broncogénico, el enfisema lobar congénito y las alteraciones vasculares.
Las enfermedades hereditarias tienen su base en una alteración de los genes y pueden afectar a las vías respiratorias, al parénquima y a las estructuras vasculares del pulmón. Incluyen enfermedades muy diversas como el déficit de alfa-1 antitripsina, el síndrome de inmotilidad ciliar o la fibrosis quística que será, por su frecuencia y relevancia clínica, la que estudiaremos en esta actualización.
Palabras clave
Congenital diseases of the respiratory system comprise a large number of embryonic development diseases that can compromise the development of the larynx, trachea, bronchi, pulmonary parenchyma, vessels, diaphragm, or thoracic wall. Prenatal ultrasound has made it possible for a large number of lung malformations to be detected before birth. Imaging techniques (radiography, ultrasound, computed tomography) are essential for diagnosis. The treatment is surgical in most cases. We will study embryology, lung development abnormalities, cystic adenomatoid malformation, sequestration, bronchogenic cyst, congenital lobar emphysema, and vascular abnormalities.
Hereditary diseases originate from genetic abnormalities. They can affect the respiratory tract, parenchyma, and vascular structures of the lung. They include very diverse diseases such as alpha-1 antitrypsin deficiency, primary ciliary dyskinesia, or cystic fibrosis which, given its frequency and clinical relevance, is the disease we will study in this update.
Keywords
Las enfermedades congénitas del aparato respiratorio comprenden un extenso número de patologías que pueden comprometer el desarrollo de la laringe, la tráquea, los bronquios, el parénquima pulmonar, los vasos, el diafragma o la pared torácica. Son entidades relativamente raras y variables en su forma de presentación, que va desde cuadros clínicos característicos a variaciones anatómicas que no requieren tratamiento y constituyen hallazgos radiológicos o endoscópicos. Mientras que en épocas anteriores se presentaban como episodios de dificultad respiratoria en el neonato o con complicaciones (infecciones, hemoptisis) o hallazgo radiológico a lo largo de la vida (secuestro, quiste broncogénico, anomalías vasculares)1. En la actualidad, los avances en la ecografía prenatal han hecho posible que un gran número de malformaciones pulmonares sean ya conocidas antes del nacimiento (segundo trimestre del embarazo), lo que ha permitido poner en duda desde algunos conceptos del desarrollo embriológico hasta la conducta terapéutica tanto prenatal como posnatal2.
Las enfermedades pulmonares hereditarias pueden afectar a las vías respiratorias, al parénquima y a las estructuras vasculares del pulmón3. Estas afecciones incluyen trastornos monogénicos simples como el síndrome de Kartagener y el déficit de alfa-1 antitripsina, en los que las mutaciones de genes críticos son suficientes para inducir fenotipos de enfermedad bien definidos como la discinesia ciliar primaria, el síndrome de Kartagener y el enfisema por déficit de alfa-1 antitripsina, y trastornos más complejos como diversas enfermedades metabólicas y multisistémicas (enfermedad de Pompe, mucopolisacaridosis, etc.). Entre todas estas enfermedades, destaca por su frecuencia y relevancia la fibrosis quística (FQ) que será la que estudiemos en esta actualización de la revista Medicine.
Enfermedades pulmonares congénitasEn esta actualización estudiaremos las anomalías con mayor relevancia clínica y diagnóstica, y para ello es fundamental conocer, en primer lugar, el desarrollo embriológico del aparato respiratorio.
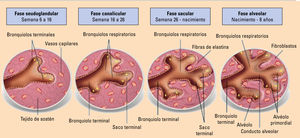
EmbriologíaEl desarrollo del pulmón se divide en las fases que se muestran en las figuras 1 y 2.

Maduración pulmonar durante el período fetal. Adaptada de Naranjo Gozalo S, et al1.

Desarrollo embrionario pulmonar. Adaptada de Naranjo Gozalo S, et al1.
Al final de la cuarta semana de gestación se forma el divertículo laringotraqueal (divertículo respiratorio) en la porción ventral de la parte caudal del intestino primitivo anterior. A medida que el divertículo se alarga, se recubre de mesénquima esplácnico y su extremo distal se elonga para formar la yema pulmonar. Esta se divide en dos yemas bronquiales que darán las ramificaciones bronquiales asimétricas (3 bronquios principales en el lado derecho y 2 en el izquierdo). En esta fase, comienza también la formación de los vasos pulmonares a partir del sexto arco faríngeo.
Fase pseudoglandular (semana 6 a 16)Durante esta fase se desarrollan las 16 subdivisiones de la vía aérea, completándose así la formación de la vía aérea conductora. Aparecen el cartílago y el epitelio ciliado. Al final de esta fase, los vasos sanguíneos pulmonares comunican con el corazón.
Fase canalicular o acinar (semana 16 a 26)Esta fase se superpone con la pseudoglandular, ya que los segmentos craneales pulmonares maduran con mayor rapidez que los caudales. Durante este período, la luz de los bronquios y bronquiolos terminales crece y el tejido pulmonar se capilariza. Así, se forman los bronquiolos respiratorios, los conductos alveolares y los sacos terminales (alvéolos primitivos) que permiten ya la respiración.
Fase sacular (semana 26 - nacimiento)A partir de la semana 26, se desarrollan muchos sacos terminales más y se crea la barrera sangre-aire al ponerse en íntimo contacto el epitelio respiratorio y el endotelio capilar, permitiéndose el intercambio gaseoso para que el feto sobreviva si nace de forma prematura. La red capilar prolifera alrededor del alvéolo y se crean capilares linfáticos. Los sacos terminales se revisten con células epiteliales escamosas llamadas células alveolares tipo I o neumocitos tipo I y con células epiteliales redondeadas secretoras o neumocitos tipo II. Estas últimas secretan el surfactante pulmonar necesario para el intercambio de gases.
Fase alveolar (del período fetal tardío hasta los ocho años)Hacia el período fetal tardío se produce un adelgazamiento de la membrana alveolocapilar (barrera pulmonar de difusión o membrana respiratoria) que permite el intercambio de gases. Los pulmones deben estar bien desarrollados para que sean capaces de funcionar en el momento del nacimiento, para ello requieren cambios de adaptación como son la producción de agente tensoactivo adecuado en los alvéolos (surfactante) y el establecimiento paralelo de las circulaciones pulmonar y sistémica. Después del nacimiento, los alvéolos primitivos crecen según se van expandiendo los pulmones y aumenta el número de bronquiolos respiratorios y alvéolos hasta los 8 años.
Este complejo proceso de desarrollo puede verse alterado en diversas formas. La separación defectuosa del brote pulmonar desde el intestino primitivo daría origen a las atresias de esófago y de tráquea y a las fisuras laringotraqueales. Una separación completa anormal sería responsable de quistes o duplicaciones bronquiales o enterógenos, secuestros extralobares, etc. La alteración de la maduración histológica dará origen a traqueomalacia, hamartomas, enfisema lobar y linfangiectasias. Las aplasias, hipoplasias y agenesias serían consecuencia de una detención de las gemaciones, mientras que la alteración del desarrollo de la circulación produciría fístulas arteriovenosas, síndrome de la cimitarra y secuestros pulmonares (figs. 1 y 2)4.
ClasificaciónEn la tabla 1 se muestra una clasificación resumida de las malformaciones pulmonares congénitas más relevantes. De esta tabla, estudiaremos las más importantes y las que presentan una mayor repercusión clínica y a la hora de establecer diagnósticos diferenciales.
Malformaciones pulmonares congénitas
| 1. Traqueales y broncopulmonaresAgenesia, aplasia e hipoplasia pulmonarSecuestro pulmonarQuiste broncogénicoBronquiectasias congénitasMalformación adenomatoide quísticaEstenosis traqueal o bronquial congénitaAtresia bronquial congénitaEnfisema lobar congénitoFístulas traqueo-esofágicas o bronco-esofágicas |
| 2. VascularesEstenosis o atresia de la arteria pulmonarAneurismas congénitos de la arteria pulmonarEstenosis o atresia de la vena pulmonarDrenaje pulmonar venoso anómaloPulmón hipogenético (síndrome de la cimitarra)Fístula arteriovenosa congénita |
| 3. DiafragmaHernia diafragmática congénita |
La agenesia y la aplasia pulmonar corresponden a la falta de desarrollo de un pulmón. En la agenesia no hay bronquio ni pulmón, mientras que en la aplasia sí existe un bronquio rudimentario, pero sin parénquima pulmonar. La agenesia se produce cuando existe un fallo del desarrollo de la yema pulmonar primitiva que condiciona una ausencia completa de bronquios, tejido pulmonar y vasos sanguíneos. La tráquea termina en un remanente de tejido pulmonar inmaduro o en un fondo de saco. Puede ser bilateral (incompatible con la vida) o unilateral. Los casos unilaterales se asocian con frecuencia con otras malformaciones congénitas, sobre todo si el pulmón que falta es el derecho. La aplasia pulmonar se caracteriza por la ausencia de parénquima pulmonar y de vasos sanguíneos pulmonares, pero se mantiene un vestigio de bronquio (fondo de saco) que es fuente continua de infecciones. La hipoplasia presenta un pulmón y unos bronquios pequeños y un escaso desarrollo vascular (tabla 2)1.
La causa es desconocida, pero se ha asociado a la trisomía 18 y, en la mayoría de los casos, cursa de forma asintomática debido al crecimiento compensatorio del otro pulmón. El diagnóstico prenatal se realiza mediante ecografía, y ya después del nacimiento mediante una radiografía de tórax efectuada por otro motivo (hallazgo casual). Su confirmación se realiza por una tomografía computarizada (TC) del tórax que permite, además, identificar otras malformaciones asociadas5,6. La espirometría muestra un patrón obstructivo debido a la distorsión y compresión del bronquio principal presente por las estructuras mediastínicas. Por último, el tratamiento médico va dirigido a la prevención y curación de las infecciones pulmonares, y el tratamiento quirúrgico a resecar el tejido residual, con el fin de evitar las infecciones respiratorias de repetición que pueden presentar estos pacientes, corrigiendo las malformaciones asociadas y las compresiones traqueobronquiales si están presentes. El pronóstico, a pesar de que existen formas asintomáticas, no es bueno, y la supervivencia de estos pacientes es pobre debido a la predisposición a las infecciones respiratorias de repetición y al gran número de malformaciones más graves asociadas, siendo las de peor pronóstico la atresia esofágica o la fístula traqueoesofágica.
Malformación adenomatoidea quísticaLa malformación adenomatoidea quística (MAQ) es la malformación congénita pulmonar más frecuente (25%). Suele ser unilateral, sin prevalencia por ninguno de los dos lados. Es más habitual localizarla en lóbulos inferiores y, en ocasiones, afecta a más de un lóbulo. La MAQ es una masa intrapulmonar multiquística causada por una proliferación de bronquiolos terminales y por la supresión del crecimiento alveolar. Los quistes grandes comunican con el árbol traqueobronquial y su vascularización depende de la circulación pulmonar. A nivel histológico, se observa una hiperplasia compensadora de las vías aéreas dístales por la falta de desarrollo de los alvéolos. Aparecen proyecciones polipoideas de la mucosa, músculo liso y tejido elástico en las paredes del quiste, con ausencia de cartílago y presencia de células secretoras de moco2.
Patogenia. La patogenia es desconocida y el origen podría ser una atresia de un bronquio segmentario por afectación embrionaria de la circulación bronquial. Una explicación podría ser que en los pacientes con MAQ se produjera una detención en la maduración durante la fase acinar y no se desarrollaran los alvéolos. Para compensar este déficit, los bronquios existentes siguen creciendo y forman dilataciones quísticas. Otra teoría es un posible aumento de la proliferación de tejido pulmonar y una disminución de la apoptosis.
Tipos. La MAQ puede ser de diferentes tipos, siendo las de tipo I y II las más frecuentes con diferencia:
Tipo 0 (1%-3%). Probable origen traqueal, quistes con tamaño inferior a 0,5cm.
Tipo I o forma macroquística (65%). Quiste único o múltiples con un tamaño mayor de 2cm.
Tipo II o forma intermedia microquística (20%-25%). Asocia malformaciones en un 60% de los casos y en ella aparecen múltiples quistes con un tamaño de 0,5-2cm.
Tipo III o forma sólida (8%). La lesión presenta un aspecto sólido, son muy grandes y en ella se combinan elementos sólidos y quistes menores de 0,5cm.
Tipo IV (2%-4%). Tiene un origen distal (alveolar o acinar) y el tamaño de los quistes es mayor de 7cm.
Clínica y diagnóstico. Clínicamente va a manifestarse como distrés respiratorio en los primeros días de vida o como infecciones pulmonares de repetición en niños. También pueden aparecer casos de neumotórax por rotura espontánea de los quistes.
El diagnóstico puede efectuarse mediante ecografía prenatal a partir de las 11-12 semanas de gestación, aunque algunos casos pueden regresar. Durante el período posnatal, el diagnóstico es radiológico mediante TC5,6.
Tratamiento. El tratamiento es quirúrgico y consiste en la resección de las zonas pulmonares afectas.
Secuestro pulmonarSon zonas de parénquima pulmonar que presentan irrigación sistémica y, en general, sin comunicación con el árbol bronquial. Los vasos arteriales pueden provenir en la gran mayoría de casos de la aorta torácica, abdominal o del tronco celíaco a través del diafragma, o bien de forma poco frecuente de vasos intercostales. Puede haber más de un vaso arterial y el retorno venoso ocurre en casi todos los casos a través de las venas pulmonares1.
Tipos. Existen 2 tipos de secuestro: el intralobar y el extralobar como muestra la tabla 31.
Diferencias entre secuestro intralobar y extralobar
| Intralobar | Extralobar | |
|---|---|---|
| Presentación | Raro en < 2 años | > 6 meses |
| Síntomas | Infecciones | Dificultad respiratoria |
| Apariencia | Pleura visceral pulmón | Pleura visceral propia |
| Localización | Lóbulos inferiores | 98% lóbulo inferior izquierdo |
| Irrigación sistémica | 80% aorta torácica o abdominal | 75% aorta torácica o abdominal |
| Drenaje venoso | 95% venas pulmonares | 80% venas sistémicas |
| Anomalías asociadas | Raro | Frecuente (> 65%) |
Secuestro intralobar. Es el más frecuente, pero suele presentarse de forma más tardía. Tiene características diferentes que le hacen ser una entidad separada, cuestionándose incluso su origen congénito. Se comporta como una zona de parénquima dentro de un lóbulo pulmonar con su pleura visceral que está desconectada de la vía aérea y recibe circulación arterial sistémica. Afecta a los lóbulos pulmonares inferiores en el 98% de los casos, con una ligera predominancia por el lado izquierdo (55%), especialmente el segmento basal posterior (fig. 3). En ocasiones puede haber pequeñas comunicaciones con el árbol traqueobronquial. Cerca de un 40% de los secuestros intralobares tienen histología semejante a una MAQ. Actualmente se plantea que los secuestros intralobares no serían congénitos, sino que la circulación arterial sería consecuencia de inflamación crónica (neovascularización).

Secuestro extralobar. Corresponde a tejido pulmonar separado del resto del pulmón y rodeado por su propia pleura, recibiendo también circulación arterial sistémica y sin conexión con la vía aérea. Es una masa de tejido pulmonar atelectásico embrionario, displásico y habitualmente quístico que dispone de envoltura pleural propia. El 80%-90% se localizan en el hemitórax izquierdo, adyacente al esófago, entre el lóbulo inferior y el diafragma. La vascularización arterial sistémica puede ser única o múltiple y, en la mayoría de los casos (80%), procede de ramas aberrantes de la aorta torácica o abdominal, pudiendo tener también otros orígenes (15%) como las arterias intercostales, la gástrica izquierda, la suprarrenal, etc. El retorno venoso es otra de las características que lo diferencian de los intralobares, al realizarse a través de la circulación general (sistema ácigos, hemiácigos, vena cava, aurícula derecha, sistema porta), aunque un porcentaje no desdeñable (25%) presenta un drenaje venoso a las venas pulmonares. Se asocia con mayor frecuencia que el intralobar a otras malformaciones congénitas (65%), en especial la hernia congénita diafragmática.
Clínica. La mayoría de los secuestros extralobares se manifiestan en forma de dificultad respiratoria en los primeros 6 meses de vida y en la niñez, siendo excepcional su diagnóstico en la edad adulta. El secuestro intralobar suele presentarse en forma de infecciones repetidas en adolescentes o adultos jóvenes, siendo raro encontrarlo en niños pequeños.
Diagnóstico. Las herramientas diagnósticas básicas para el diagnóstico del secuestro son la ecografía prenatal, la radiografía de tórax, la angio-TC y la arteriografía6. El secuestro extralobar se diagnostica cada vez con mayor frecuencia de forma prenatal como una masa de hemitórax izquierdo que suele disminuir de tamaño con el progreso de la gestación. La forma más frecuente de presentación del secuestro corresponde a la de una masa densa basal izquierda, cercana a la columna en una radiografía de tórax. Una vez que se sospecha el diagnóstico, se debe tratar de identificar la circulación arterial mediante TC vascular o arteriografía. Radiológicamente se presenta con zonas de condensación o áreas quísticas infectadas con niveles hidroaéreos en su interior. Clínicamente se manifiestan como infecciones repetidas, hemoptisis o síntomas inespecíficos de disnea y cansancio fácil.
Tratamiento. El tratamiento del secuestro pulmonar es quirúrgico, e idealmente ha de realizarse antes de que aparezcan complicaciones. Hay que tener en cuenta la infección como la más frecuente y recordar que algunos secuestros tienen histología tipo MAQP y son susceptibles de malignización (fig. 3).
Quistes broncógenosSon lesiones quísticas pulmonares o mediastínicas revestidas por epitelio columnar ciliado de tipo bronquial. Se desarrollarán a partir de una gemación anómala del intestino primitivo. Si esta separación ocurre de forma precoz, el quiste permanece en el mediastino y, si ocurre de forma tardía, da origen a los quistes periféricos. Los quistes poseen una pared propia delgada con cartílago, musculatura lisa y glándulas bronquiales. Pueden tener comunicación con la vía aérea y, si la formación del quiste fue muy precoz, pueden existir zonas con epitelio gástrico o esofágico. Los quistes suelen ser redondeados y uniloculares, y el contenido puede ser aéreo, seroso o mucoso. A menudo son únicos, y gran parte de las publicaciones encuentran una mayor frecuencia de ubicación mediastínica y de preferencia al lado derecho. Se ubican a lo largo de la tráquea, cerca de la carina o del hilio entre la vía aérea y el esófago1,2.
Clínica y diagnóstico. La mayoría de los quistes broncógenos aparecen como un hallazgo radiológico, ya sea por una imagen de ensanchamiento mediastínico o por la aparición en una imagen quística pulmonar única. Cuando presentan sintomatología, esta puede corresponder a la hiperinsuflación del quiste con la compresión del tejido circundante o a la infección de la cavidad quística. La TC es la prueba principal que confirma el diagnóstico y define las características de la lesión5.
Tratamiento. El tratamiento debe ser quirúrgico para evitar las complicaciones que incluyen, al igual que en la MAQ y en los secuestros, la infección y la malignización.
Enfisema lobar congénitoEs una sobredistensión de uno o más lóbulos que son histológicamente normales. No hay destrucción del parénquima sino atrapamiento aéreo debido a un mecanismo valvular. La causa no es conocida, pero puede ser un efecto valvular bronquial sobre los lóbulos afectos y que da lugar a un atrapamiento aéreo e hiperinsuflación. Se han descrito otras causas para explicar esta alteración, como la falta de un adecuado soporte cartilaginoso bronquial3.
Se suele presentar como un cuadro de distrés respiratorio en niños menores de un año de vida, por lo que es raro verlo en adultos. El cuadro respiratorio puede ser muy grave y, en los casos que no se manifiestan de forma precoz, suele producirse un retraso del crecimiento e infecciones pulmonares de repetición. El diagnóstico es radiológico, apreciándose la hiperinsuflación que, en la mayoría de las ocasiones, se produce en los lóbulos superiores y en un 20% es bilateral.
El tratamiento es la resección quirúrgica en los casos en los que la enfermedad se halla localizada. Cuando hay afectación difusa, no se debe intervenir y debe tenerse en cuenta la posible existencia de malformaciones asociadas, sobre todo cardíacas.
Malformaciones vascularesEn la tabla 1 se recoge una amplia relación de alteraciones de los vasos pulmonares, tanto a nivel arterial como venoso y linfático. De ellas estudiaremos las fístulas arteriovenosas y el síndrome de la cimitarra.
Fístulas arteriovenosas pulmonaresPueden presentarse en forma aislada o en el seno del síndrome de Rendu-Osler-Weber (telangiectasia hereditaria familiar). La circulación arterial de la fístula viene de la arteria pulmonar, aunque también puede hacerlo de la circulación sistémica; puede ser única o múltiple, incluso con compromiso bilateral. Dependiendo del tamaño de la fístula se presenta clínicamente con disnea, cianosis, acropaquías y hemoptisis. La sospecha diagnóstica se confirma mediante angio-TC y arteriografía para precisar con exactitud el tamaño y la ubicación de la fístula. El tratamiento puede ser quirúrgico o mediante embolización, dependiendo de las características de la fistula y de la situación funcional del paciente5,6.
Síndrome de la cimitarraEs una alteración de la circulación venosa pulmonar. Usualmente afecta al pulmón derecho que es hipoplásico y cuya circulación venosa drena a través de un vaso largo hasta la cava inferior. Esto da en la radiografía una imagen que semeja una cimitarra. Puede tener zonas con circulación arterial sistémica y puede acompañarse de pulmón en herradura o secuestros pulmonares. El tratamiento es la corrección quirúrgica.
Mensajes para recordarLos diversos tipos de malformaciones congénitas expuestas tienen características comunes, y su conocimiento puede ayudar a un correcto abordaje de las mismas1.
- 1.
Las alteraciones se manifestarán principalmente en el recién nacido o durante la infancia, siendo más rara su expresión en la edad adulta, salvo los casos en los que no se produzca sintomatología y sea un hallazgo radiológico.
- 2.
Hay que pensar siempre en la posibilidad de que existan otras malformaciones congénitas asociadas.
- 3.
Deben estar presentes en la sospecha diagnóstica, ante un neonato con clínica de distrés respiratorio, o ante niños o adultos jóvenes con clínica respiratoria constante e infecciones respiratorias recurrentes, dado que esta clínica suele ser bastante común en casi todas ellas.
- 4.
Si se produce un diagnóstico prenatal es importante iniciar un seguimiento ecográfico estricto, complementándolo con otras pruebas como el Doppler o la resonancia magnética (RM) prenatal que permitan realizar un diagnóstico diferencial. Este control podrá mostrar si existe una evolución hacia la desaparición o la estabilidad o si se produce un agravamiento que pueda llegar a requerir maniobras terapéuticas en el feto.
- 5.
Existen diferentes recursos diagnósticos, pero la mayoría podrán ser diagnosticadas por radiología simple de tórax, TC y RM.
- 6.
Es importante conseguir inicialmente una estabilidad clínica del paciente que permita un estudio completo y exacto.
- 7.
Es fundamental contar con un equipo multidisciplinar que permita abarcar los diferentes aspectos que engloban.
Las enfermedades hereditarias se heredan de los padres, es decir, tienen una base en una alteración de los genes, mientras que las congénitas aparecen desde el nacimiento como consecuencia de alteraciones del desarrollo embrionario. Una mayor comprensión de la base genética de las afecciones pulmonares ha proporcionado nuevos conocimientos sobre su fisiopatología y ha ayudado en algunos casos a arrojar luz sobre formas esporádicas más comunes. Es importante destacar que la identificación de genes causantes también ha permitido el diagnóstico prenatal y el asesoramiento genético para muchas enfermedades3. En este apartado estudiaremos la FQ, que es la enfermedad genética más frecuente que afecta al aparato respiratorio.
Fibrosis quísticaLa FQ es una enfermedad multisistémica hereditaria que afecta a las glándulas exocrinas. Puede afectar al aparato digestivo, a las glándulas sudoríparas y al aparato reproductor, entre otros órganos y sistemas, pero la mayor morbimortalidad viene determinada por el progresivo deterioro del pulmón7. La FQ se transmite con carácter autosómico recesivo, y es una de las enfermedades hereditarias más prevalentes en la raza blanca, con una prevalencia que se sitúa entre los 6-8 casos por 100000 habitantes.
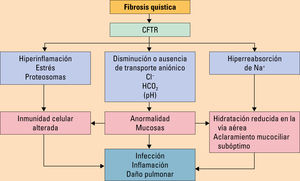
EtiopatogeniaLa FQ está causada por mutaciones en un gen localizado en el brazo largo del cromosoma 7 (7q31) que se transmite con herencia autosómica recesiva. La frecuencia de las distintas mutaciones es diferente en función del área geográfica analizada. En la población de raza blanca, la primera mutación descrita y la más frecuente es la F508del. La enfermedad se caracteriza por una gran variabilidad en su expresión, debida posiblemente a que más de 1000 mutaciones en la secuencia de este gen están asociadas a manifestaciones clínicas. Este gen codifica la proteína reguladora de la conductancia transmembrana (CFTR) que es una glucoproteína localizada en la membrana apical de las células epiteliales y funciona como un canal de cloro regulado por AMP cíclico. La alteración del canal CFTR afecta al transporte de cloro y bicarbonato y, como consecuencia, se produce un transporte anormal de electrolitos y agua a través de la membrana apical de los epitelios afectados (se bloquea la absorción de cloro y aumenta la reabsorción de sodio, incrementando pasivamente la reabsorción de agua)8. Existen cuatro grandes grupos de defectos en el gen de la CFTR que generan producción, procesamiento, regulación y conducción defectuosos de la proteína.
La patogénesis no es del todo conocida, pero las alteraciones en la CFTR causan un mal funcionamiento en diferentes canales para la conducción de iones en todos los tejidos exocrinos, provocando secreciones espesas difíciles de movilizar y produciéndose un círculo vicioso obstrucción-inflamación-infección en el aparato respiratorio. En el pulmón, lleva a la deshidratación de la capa de líquido de superficie de la vía aérea y a la alteración del aclaramiento mucociliar. Estos problemas secretorios son una característica fundamental de la enfermedad, ya que los tapones mucosos y la dilatación de los ductos y túbulos de las glándulas submucosas están presentes incluso en neonatos. Con el tiempo, la secreción persistente de moco junto con la disminución del líquido periciliar provoca un moco deshidratado y viscoso que obstruye las vías aéreas y favorece la infección9,10.
Es importante resaltar la importancia de los mecanismos inflamatorios que permiten la invasión bacteriana, a través de los epitelios dañados, por microrganismos específicos típicos de la FQ, como son Staphylococcus aureus, Haemophilus influenzae y Pseudomonas aeruginosa que, a su vez, estimulan la respuesta inflamatoria mediada por neutrófilos, incrementando aún más el daño tisular.
ClínicaLa clínica respiratoria de la FQ está caracterizada por la presencia de tos productiva, hiperinsuflación de ambos campos pulmonares en la radiografía de tórax y pruebas de función respiratoria con una alteración de tipo obstructivo.
La evolución natural de la enfermedad se ve influencia por la aparición de exacerbaciones caracterizadas por clínica infecciosa de disnea y, sobre todo, incremento de la tos con cambio en las características del esputo (volumen, color, viscosidad) (fig. 4).
. Modificada de Olveira C, et al10.")
. Efectos de la disfunción de la proteína reguladora de la conductancia transmembrana (CFTR). Modificada de Olveira C, et al10.
El diagnóstico se basa en la presencia de características fenotípicas compatibles o la historia de enfermedad en familiares (hermanos o primos), junto con una prueba de laboratorio que evidencie una disfunción de la CFTR (tabla 4, fig. 1).
Criterios diagnósticos de la fibrosis quística
| 1. Uno o más criterios clínicosCaracterísticas fenotípicas consistentesPoliposis nasal y/o alteraciones de los senos paranasalesSíndromes pierde sal: pérdida aguda de sal, alcalosis metabólica crónicaAlteraciones gastrointestinales y nutricionalesIntestinal: íleo meconial, obstrucción intestinal distal, prolapso rectalPancreática: insuficiencia pancreática, pancreatitis recurrente Hepática: hepatopatía crónica sugerida por el cuadro clínico o por características histológicas de cirrosis biliar focal o multilobular Nutricional: retraso de crecimiento (malnutrición proteico-calórica), hipoproteinemia y edema, complicaciones secundarias a déficit de vitaminas liposolublesAlteraciones urogenitales en el hombre: azoospermia obstructivaHistoria familiar consistenteCribado neonatal positivo |
| 2. Una o más pruebas que evidencien disfunción de la proteína reguladora de la conductancia transmembrana CFTRCloro en sudor > 60 mmol/l en dos ocasionesDetección de dos mutaciones de fibrosis quística en diferentes alelosAlteración de la diferencia de potencial nasal |
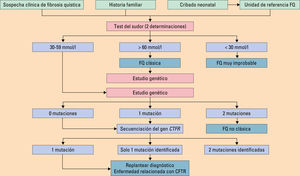
Prueba del sudor. Sigue siendo la prueba inicial fundamental en el diagnóstico de la FQ, dado que puede identificar casi el 90% de las mutaciones más frecuentes. La determinación del cloro en el sudor es el método bioquímico para confirmar el diagnóstico de FQ. Se considera positiva una concentración de cloro superior a 60 mmol/l; negativa, una concentración de cloro menor de 30 mmol/l y dudosa entre 30-59 mmol/l. Una concentración de cloro en sudor mayor de 60 mmol/l distingue a la mayoría de los pacientes con FQ. La prueba debe realizarse, al menos, dos veces en días diferentes. Una prueba negativa no excluye el diagnóstico y, si existe una elevada sospecha, hay que realizar un estudio genético9,10.
Diagnóstico molecular. Para hacer el diagnóstico de la FQ mediante estudio molecular es necesario identificar una mutación del gen CFTR en cada uno de los alelos del paciente. Se realiza por análisis directo de la mutación mediante técnicas que identifican las mutaciones conocidas en el gen de la CFTR. Estas técnicas identifican casi el 90% de las mutaciones más comunes. Para realizar el diagnóstico se precisa determinar la existencia de, al menos, dos mutaciones causantes de alteración en la función de la CFTR7,8.
Diferencia de potencial nasal. Las anormalidades del transporte iónico en el epitelio respiratorio del paciente con FQ están asociadas con un potencial eléctrico diferente al de los individuos sanos. En el epitelio respiratorio se produce un transporte de iones Cl y Na que genera una diferencia de potencial transepitelial que es diferente entre personas sanas y enfermas con FQ. En estos, se observa un patrón de anormalidad como consecuencia del aumento en la reabsorción de sodio, que lo hace más electronegativo, existiendo una gran diferencia con los valores obtenidos en la población sana (-46mV en FQ/-19mV en población sana). En la actualidad no es muy utilizado, pues no está disponible en la mayoría de los centros y es una prueba difícil de estandarizar9,10.
Cribado neonatal. El cribado neonatal de la FQ ha permitido el diagnóstico precoz de la enfermedad, siendo determinante en el aumento de la supervivencia de estos pacientes. Su principal inconveniente es su baja especificidad y su elevado número de falsos positivos. Los niveles en suero de tripsina inmunorreactiva (TIR) son más altos en los recién nacidos con FQ que en aquellos que no están afectados por la enfermedad. La determinación de la TIR en una gota de sangre seca es el método empleado y está contemplada su realización, en la mayoría de los protocolos actuales, entre el tercer y quinto día de vida. La concentración elevada de TIR en sangre es consecuencia de la obstrucción parcial o completa de los canalículos pancreáticos que produce que refluya dicha enzima hacia la circulación sanguínea. Las mediciones de dicha enzima, junto con la prueba del sudor y el análisis de las mutaciones, constituyen la base de los programas de detección selectiva de FQ en los recién nacidos9,10.
El pulmón en la fibrosis quísticaLa importante morbimortalidad de la FQ está relacionada fundamentalmente con la afectación pulmonar y sus complicaciones, condicionando el pronóstico. La alteración de la proteína CFTR provoca cambios en la reología del moco y en el aclaramiento mucociliar y una reacción inflamatoria exagerada que se localiza fundamentalmente en la vía aérea. Las secreciones obstruyen las vías aéreas y alteran la eliminación de microorganismos potencialmente patógenos, favoreciendo la infección crónica. Esta infección-inflamación se asocia con un daño progresivo con inflamación neutrofílica, desarrollo de bronquiectasias, neumopatía crónica progresiva con destrucción del parénquima pulmonar, deterioro de la función pulmonar e insuficiencia respiratoria7.
Las pruebas de imagen (TC, RM), la exploración funcional pulmonar y los estudios microbiológicos son las herramientas básicas para el diagnóstico y seguimiento de la afectación respiratoria en la FQ8–10. La TC de tórax es la prueba clave para conseguir el diagnóstico precoz de la afectación pulmonar en la FQ, y también es útil, y con más precisión que las pruebas funcionales, para estimar la gravedad y la progresión de la afectación pulmonar, ayudando a valorar la respuesta al tratamiento y a tomar decisiones sobre nuevos tratamientos, ajustándolos a la gravedad de las lesiones. Una progresión rápida del atrapamiento aéreo y de las bronquiectasias en la TC se ha asociado con la presencia de infección pulmonar y con peor pronóstico. La exploración de la función respiratoria muestra la existencia de un patrón obstructivo que va progresando en el tiempo, y que asocia la existencia de atrapamiento aéreo e hiperinsuflación pulmonar.
La colonización, la infección crónica y las exacerbaciones repetidas son una característica constante de la enfermedad, de aquí la importancia de los estudios microbiológicos. En las fases iniciales es característica la presencia, en los cultivos de esputo, de H. influenzae y de S. aureus sensible a meticilina. Posteriormente, la mayoría de los pacientes presentan P. aeruginosa que es la bacteria más característica de la FQ. La infección bronquial crónica por P. aeruginosa es la causa más importante de morbimortalidad en los pacientes con FQ. La muestra de elección para realizar estudios microbiológicos es el esputo. En caso de que el paciente no pueda expectorar, se pueden utilizar esputos inducidos y aspirados bronquiales, reservándose el lavado broncoalveolar (broncoscopia) para pacientes con mala evolución. En los enfermos sin evidencia de infección se recomienda, al menos, un cultivo trimestral de esputo. En la infección crónica, el cultivo debe realizarse siempre que se produzcan exacerbaciones y, al menos, una vez cada 3 meses10.
El tratamiento antibiótico debe iniciarse en función del cultivo y del antibiograma, y en la FQ existe indicación de tratamiento antibiótico inhalado en los casos de infección crónica. Los fármacos inhalados se depositan directamente en el tracto respiratorio, con lo que se alcanzan altas concentraciones, con un inicio de acción más rápido y con menores efectos secundarios que si se emplea la vía sistémica. Las concentraciones séricas alcanzadas por los antibióticos tras su administración inhalada son muy escasas y, por ello, tienen escasa toxicidad. El desarrollo tecnológico de los últimos años ha permitido contar con dispositivos que optimizan el depósito pulmonar y disminuyen el tiempo necesario para realizar el tratamiento. Debido a que con la administración por vía inhalada se alcanzan elevadas concentraciones del antibiótico en la vía aérea, la elección del antibiótico se realizará según el microorganismo que cause la infección, pero no según las sensibilidades del antibiograma convencional.
Como tratamientos no antibióticos, se dispone de diferentes sustancias que mejoran el aclaramiento mucociliar9,10. La dornasa ? (rhDNasa) (Pulmozyme®) es una solución altamente purificada de deoxirribonucleasa humana recombinante que reduce la viscosidad del moco al digerir el ADN extracelular secretado por el neutrófilo. El manitol es un polialcohol que, por su capacidad osmótica, mejora el aclaramiento mucociliar. El suero salino hipertónico al 7% nebulizado aumenta el volumen del líquido periciliar e induce tos por el impacto de la sal en la faringe, por lo que mejora el sistema de aclaramiento mucociliar, favoreciendo el drenaje de secreciones. Existe una formulación de suero hipertónico con ácido hialurónico que minimiza la tos, las molestias faríngeas y el sabor salado, por lo que tiene mejor tolerancia.
La rehabilitación respiratoria, el drenaje de secreciones y el ejercicio físico deben estar siempre presentes en el tratamiento de estos enfermos. Las técnicas de drenaje de secreciones favorecen la eliminación de las secreciones de las vías aéreas, mejorando la ventilación y la mecánica pulmonares, y reducen las infecciones respiratorias. En pacientes evolucionados, existe una neumopatía crónica progresiva con destrucción del parénquima pulmonar, deterioro progresivo de la función pulmonar e insuficiencia respiratoria. A medida que va disminuyendo la función pulmonar, se produce hiperinsuflación, discordancia entre la perfusión y la ventilación, disminución de la fuerza de la musculatura respiratoria e hipoventilación alveolar progresiva que conduce a hipoxemia y, finalmente, a hipercapnia. Si el paciente presenta insuficiencia respiratoria hipoxémica diurna o hipertensión pulmonar, se le debe indicar oxigenoterapia continua para evitar o retrasar la situación de cor pulmonale. La ventilación mecánica no invasiva puede ser útil en pacientes con FQ, aunque todavía los criterios de inicio no están bien establecidos10.
A pesar de la mejoría en la esperanza de vida de los pacientes afectos de FQ, la patología pulmonar sigue siendo la causa de mortalidad en el 80% de los casos, por lo que el trasplante pulmonar representa la única alternativa con impacto pronóstico positivo en la supervivencia y la calidad de vida en los estadios finales de la enfermedad. Los resultados de los pacientes con FQ trasplantados han mejorado rápidamente y la supervivencia media se acerca o incluso supera los 10 años (fig. 5).

Algoritmo diagnóstico de la fibrosis quística. Modificada de Olveira C, et al10.
Los autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
-
 Importante
Importante
-
 Muy importante
Muy importante
-
 Metaanálisis
Metaanálisis
-
 Ensayo clínico controlado
Ensayo clínico controlado
-
 Epidemiología
Epidemiología
-
 Artículo de revisión
Artículo de revisión
-
 Guía de práctica clínica
Guía de práctica clínica


