
Los errores congénitos del metabolismo (ECM) son un grupo variado de trastornos genéticos que implican anormalidades en los procesos bioquímicos celulares, cuyas consecuencias patológicas pueden aparecer a cualquier edad.
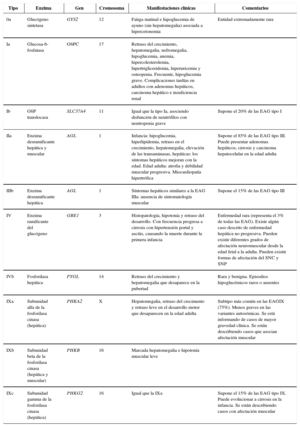
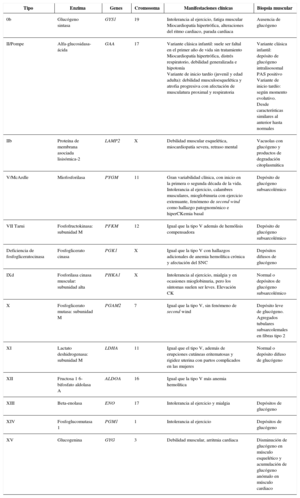
Etiopatogenia y clasificaciónLa mayoría de los ECM son causados por defectos enzimáticos, que dan como resultado una conversión inadecuada o ausente de sustratos en productos. Se heredan casi todas con un patrón autosómico recesivo. En muchos casos, los problemas surgen debido a la acumulación de dichas sustancias. Un grupo heterogéneo de los mismos tiene en común el almacenamiento de glucógeno (glucogenosis). También puede tratarse de trastornos del metabolismo de monosacáridos, como la fructosa y la galactosa.
Manifestaciones clínicasAunque la clínica suele ser multisistémica, la localización predominante de los depósitos de glucógeno determina su sintomatología. Esta puede ser fundamentalmente hepática o muscular, con afectación del músculo estriado y el corazón.
DiagnósticoEl diagnóstico tras la sospecha clínica se basa en la demostración de un déficit de actividad enzimática y el hallazgo de mutaciones en los genes correspondientes.
TratamientoTodas ellas requieren un manejo multifactorial y para algunas se han desarrollado tratamientos de sustitución enzimática.
Palabras clave
Inborn errors of metabolism (IEM) are a varied group of genetic disorders producing aberrations in cellular biochemistry and showing its clinical consequences in any age group.
Pathogenesis and classificationMost IEM are a consequence of enzyme deficiencies. As consequence, the conversion of substrates to products is inaccurate or absent. The pattern of inheritance is, usually, autosomal recessive. In many cases, clinical manifestations are caused by an accumulation of substances. Heterogeneous group of them is characterized by disturbances in glycogenesis (glycogen storage). In other cases, the disorder is placed in monosaccharide metabolism (for example: fructose and galactose).
Clinical manifestationsOften the clinical feature is multi-system, however symptomatology is determined by location of glycogen stores. There are two predominant locations for glycogen stores: liver and muscles, involving striated muscle fiber (even heart muscle)
DiagnosisBehind clinical suspicion, the detection of gene mutations related with the evidence of a low-enzyme activity will allow us to reach a diagnosis.
TreatmentA multifactorial treatment is required for all of them. Enzyme replacement therapy has been developed for only some glycogen storage diseases.
Keywords
Identifíquese
¿Aún no es suscriptor de la revista?

Comprar el acceso al artículo
Comprando el artículo el pdf del mismo podrá ser descargado
Teléfono para incidencias
De lunes a viernes de 9h a 18h (GMT+1) excepto los meses de julio y agosto que será de 9 a 15h