
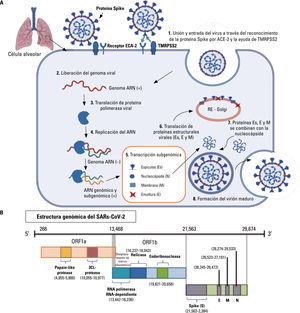
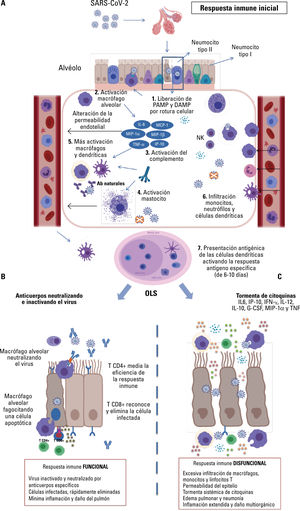
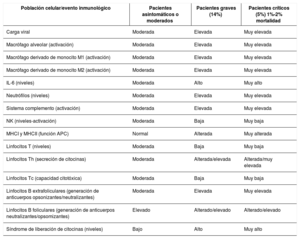
El sistema inmune es capaz de controlar adecuadamente la infección por el SARS-CoV-2 en un 81% de los pacientes, cursando de una forma asintomática o con sintomatología moderada; sin embargo, un 19% de los pacientes infectados sufren una enfermedad grave, que llega a convertirse en crítica y mortal. Este trabajo de revisión pretende proporcionar un repaso a los antecedentes epidemiológicos de los β-coronavirus, describir el mecanismo de infección del SARS-CoV-2 y resumir la base inmunológica racional que se conoce hasta la actualidad para permitir una mejor comprensión de la inmunopatología de la COVID-19. El virus SARS-CoV-2 es capaz de alterar profundamente el comportamiento de los componentes moleculares y celulares del sistema inmune. Las decisiones iniciales del sistema inmune innato son responsables de una correcta o inadecuada respuesta del sistema inmune adaptativo y, junto con las comorbilidades, están directamente asociadas a la progresión de la patología.
Palabras clave
The immune system is capable of adequately controlling SARS-CoV-2 infection in 81% of patients, whose disease is asymptomatic or who experience moderate symptoms. However, 19% of infected patients develop severe disease which can become critical or fatal. This review article intends to provide an overview of the epidemiological antecedents of β-coronaviruses, describe the mechanisms of SARS-CoV-2 infection, and summarize the rational immunological underpinnings known at present which allow for a better understanding of the immunopathology of COVID-19. The SARS-CoV-2 virus is capable of profoundly altering the behavior of molecular and cellular components of the immune system. The initial decisions of the innate immune system are responsible for a proper or improper response of the adaptive immune system and, along with comorbidities, are directly associated with disease progression.
Keywords
Identifíquese
¿Aún no es suscriptor de la revista?

Comprar el acceso al artículo
Comprando el artículo el pdf del mismo podrá ser descargado
Teléfono para incidencias
De lunes a viernes de 9h a 18h (GMT+1) excepto los meses de julio y agosto que será de 9 a 15h